
Source for feature photo: Welcome to the Future by Geralt from Pixabay
The development of massively parallel sequencing is changing the way scientists determine DNA profiles. MPS technology overcomes the limitations of current methodology, known as capillary electrophoresis. However, current MPS procedures are labor intensive and require high quality DNA, which is not realistic when analyzing samples found at crime scenes. Recently, Dr. Dongming Yang at Beijing Institute of Biomedicine led a team to develop a protocol eliminating the need for DNA extraction. By eliminating the need for extraction, they increased the DNA available for testing, reduced time needed to generate a DNA profile, and gave a more discriminating profile than what is seen with current methods.
DNA analysts most commonly measure STRs to generate a DNA profile of an individual. After an analyst extracts the DNA, quantifies the amount present, and amplifies certain regions of the genetic sequence known as STRs outlined in a previous ForensicBites article, the STRs are identified using capillary electrophoresis (CE). STRs are fragments of DNA made of short repeat sequences of nucleotides (such as TGATGATGA) that are unique to each individual. CE distinguishes STR fragments from the colored tags attached to them. However, most commercial kits for CE contain a limited number of colors, ranging between four and six, which makes differentiating fragments of similar sizes very difficult. If the scientist analyzes a sample that contains donations from multiple people, known as multi-source profile, and the STR fragments are similar sizes in the same colors, this could make the profile look like it originated from one person, known as a single source profile.
To overcome the obstacles from limited color selection overlapping on similar-sized fragments, scientists turned to a new method: massively parallel sequencing. Massively parallel sequencing (MPS) breaks down STRs to the individual nucleotide level, looking at the sequence of A, C, T and G that create the fragment’s sequence. CE determines the length of STRs by considering the fragment as a whole molecule, analogous to a single word – but MPS looks at the “letters” that make up that word. The MPS method considers the amplified fragments, sorts and rearranges the “letters” back into words based on a reference, in this case, the known sequences of the human genome, so that we know which STRs are being analyzed (Fig 1).
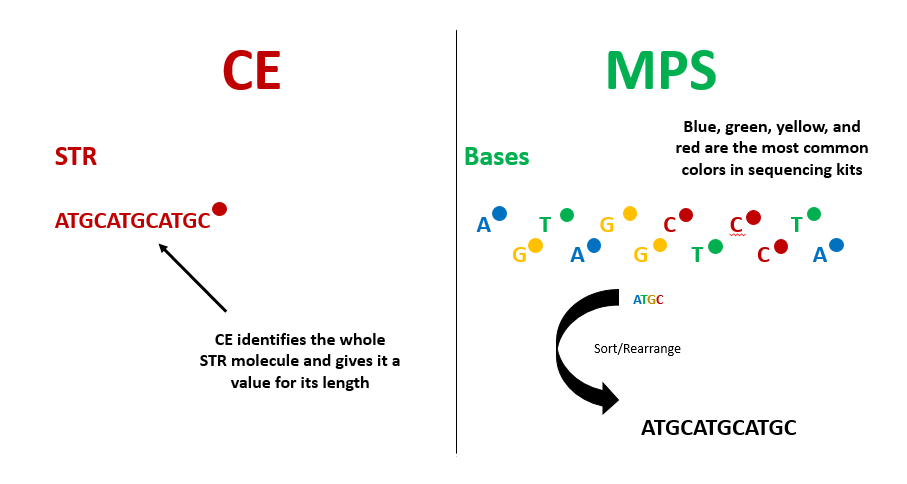
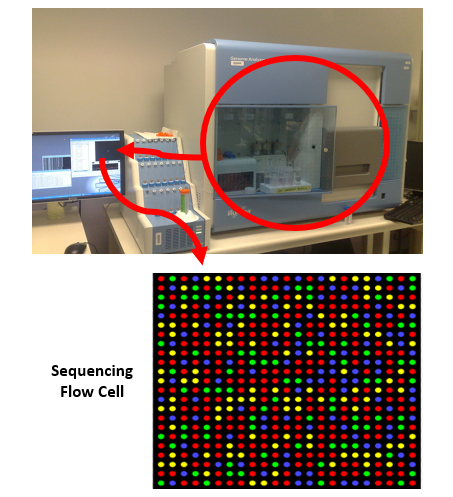
MPS eliminates the limitations of challenging analyses based on fragment sizes by measuring individuals bases. Fragments are added to a flow cell where each of the four nucleotides (A, G, T or C) is assigned a color. The machine reads the sequences by capturing the light that each individual nucleotide gives off and the system records the different combinations of the four colors detected after many cycles of amplification. Whereas CE can only observe one STR at a time, MPS can observe many STR sequences at once, increasing the output of data while decreasing the time it takes to generate a NA profile (Fig 2).
The authors first showed in their study that their MPS-based method distinguishes a greater number of alleles than the current CE-based method employed by forensic laboratories (Fig 3). MPS also demonstrated higher discrimination power between alleles. For example, a T to C substitution was found in the first repeat of CSF1PO allele 11 that was not found when using CE, and the number of alleles increased from 10 to 11 using MPS.

Then Dai et al. further optimized their system by eliminating the need for DNA extraction. By eliminating this step, the time for direct library preparation, where the adheres directly to the flow cell, decreased by 4 hours and ready for sequencing in one business day. DNA extraction allows better identification of the sample, however among all the samples that were observed in the study, the group was able to successfully identify over 90% of loci (location of the STRs of interest) using the enhanced MPS technology.
As an analyst, the most important aspect when interpreting DNA profiles is that sequencing (detection of STRs) is correct. You want to make sure you are identifying the correct individual(s). Dai et al. not only showed that sequencing via MPS can prove faster than the traditional methods of CE, but also that sequencing can accurately identify individuals and mixtures. MPS is still costly and time consuming compared to CE, and the primers used in this study need tweaking for specificity. However, with this advancement, scientists will generate DNA profiles quickly and efficiently, increasing turn-around time for current and cold cases.
| Title | High polymorphism detected by massively parallel sequencing of autosomal STRs using old blood samples from a Chinese Han population |
| Authors | Wenshen Dai, Yajiao Pan, Xiaochen Sun, Riga Wu, Luo Li & Dongming Yang |
| Journal | Scientific Report |
| Publisher | Nature |
| Year | 2019 |
| Link | https://www.nature.com/articles/s41598-019-55282-9#citeas |

{kind=link}
{kind=link}
One thought on “Back to the Future: Advances in DNA Analysis Using Massively Parallel Sequencing”